Research of the Dellago Group
Computational Statistical Mechanics
Using the computational and theoretical tools of modern statistical mechanics, we study phenomena occurring in complex ordered and disordered systems at the nanoscale. Current research focuses on stochastic thermodynamics, nanoscale materials, interfaces, phase transitions and self-assembly in soft matter. A major research effort of the group consists in developing trajectory-based sampling methods and machine learning approaches for the simulation of molecular systems.
Rare event simulation
Rare event simulation
Many processes occurring in nature of technology are characterized by wide ranges of time scales. For instance, micrometer sized water droplets can remain liquid for several milliseconds down to more than 40 degrees below freezing and at higher temperatures the lifetime of undisturbed supercooled water is essentially unlimited. Thus, the time scale for the homogeneous freezing of water typically lies many orders of magnitudes beyond the picosecond time scale of basic molecular motions. Such vast separations of time scales, originating in energetic and/or entropic barriers that obstruct the motion of the system through phase space, are a challenge for the computer simulator. In the past decades one important research effort of our group was the development of trajectory base simulation methods to sampled analyze the mechanism and kinetics of rare events.
C. Dellago, Peter G. Bolhuis, and Phillip L. Geissler, “Transition Path Sampling”, Adv. Chem. Phys. 123, 1 (2002).
E. E. Borrero and C. Dellago, “Avoiding traps in trajectory space: metadynamics enhanced transition path sampling”, Eur. Phys. J. ST 225, 1609-1620 (2016).
G. Menzl, A. Singraber, and C. Dellago, “S-shooting: a Bennett-Chandler method for the calculation of reaction rate constants from committor trajetories”, Farad. Disc. 195, 345-364 (2016).
L. Qin, C. Dellago, and E. Kozeschnik, “An efficient method to reconstruct free energy profiles for diffusive processes in transition interface sampling simulations”, J. Chem. Phys. 150, 094114 (2019).

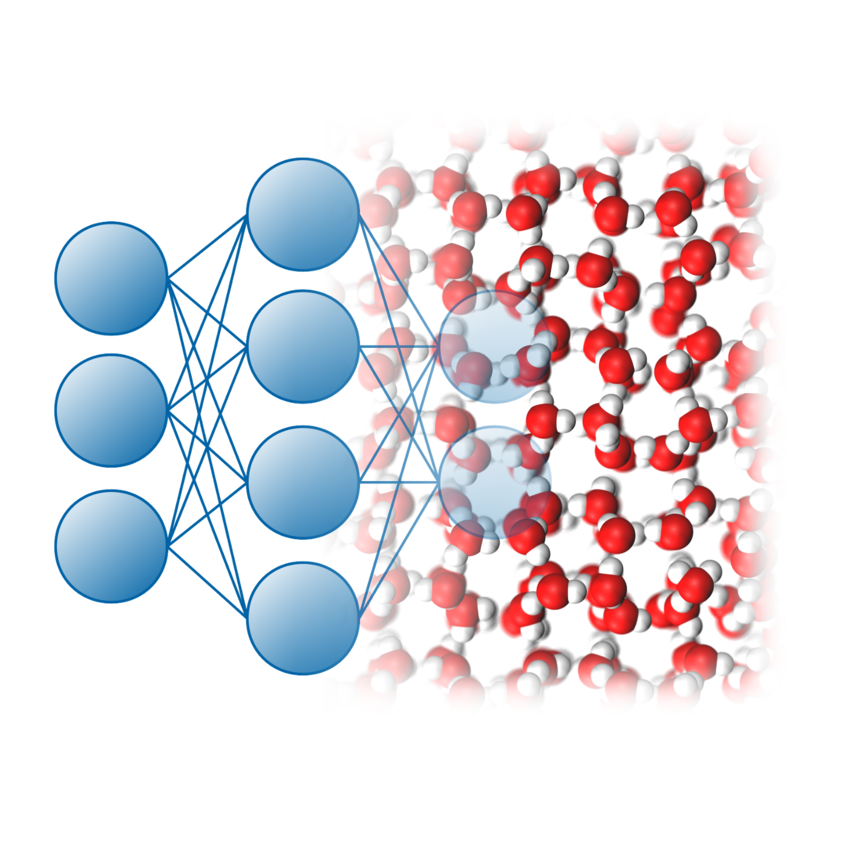
Machine learning for molecular simulation
Dynamical processes occurring in condensed matter systems can be modeled atomistically using first principles, but such calculations are very demanding, severely limiting accessible systems sizes and simulation times. Machine learning methods have recently emerged as a promising approach to address these challenges and to bridge the gap between ab initio calculations and large-scale molecular dynamics simulations of complex systems. We have recently developed a software package for the development and application of NNs, called n2p2, and have used it to create accurate ab initio quality potentials for water and other substances. Furthermore, we explore the application of machine learning methods for the analysis and rationalization of simulation results.
P. Geiger and C. Dellago. “Neural networks for local structure detection in polymorphic systems”, J. Chem. Phys. 139, 164105 (2013).
T. Morawietz, A. Singraber, C. Dellago, and J. Behler, „How Van der Waals interactions determine the unique properties of water“, Proc. Natl. Acad. Sci. USA 113, 8368-8373 (2016).
A. Singraber, J. Behler, and C. Dellago, “Library-based LAMMPS implementation of high-dimensional neural network potentials”, J. Chem. Theo. Comp. 15, 1827 (2019).
A. Singraber, T. Morawietz, J. Behler, and C. Dellago, „Parallel multistream training of high-dimensional neural network potentials”, J. Chem. Theo. Comp, 15, 3075-3092 (2019).
N2P2 on github gttps://github.com/CompPhysVienna/n2p2
Phase transitions and nucleation processes
Not too far from coexistence, first order phase transitions such as the freezing or condensation proceed by a mechanism of nucleation of growth. A qualitative picture of this process is provided by classical nucleation theory, which asserts that the transformation proceeds via the formation of a localized nucleus of the stable phase growing in the metastable phase. Due to the free energetic cost creating an interface between the two phases, the free energy of this process displays a barrier that prevents the rapid transformation to the stable phase. While classical nucleation theory provides a provides a rough picture on the nucleation process, it usually fails on a quantitative level. In our group, we use rare event simulation methods to study the thermodynamics, kinetics and microscopic mechanism of nucleation processes. Recent work has focused on cavitation of water under negative pressures and the freezing of supercooled water.
S. Jungblut and C. Dellago, “Crystallization on pre-structured seeds“, Phys. Rev. E, 87, 012305 (2013).
G. Menzl, M.A. Gonzalez, P. Geiger, F. Caupin, J.L.F. Abascal, C. Valeriani, and C. Dellago, “Molecular mechanism of cavitation in water under tension”, Proc. Natl. Acad. Sci. USA 113, 13582-13587 (2016).
B. Cheng, C. Dellago, and M. Ceriotti, “Theoretical prediction of the homogeneous ice nucleation rate: disentangling thermodynamics and kinetics”, Phys. Chem. Chem. Phys. 20, 28732-28740 (2018).

Stochastic thermodynamics
As one of the supreme achievements of 19th century science, classical thermodynamics is a cornerstone of physics and provides a general framework to understand the transformation of macroscopic matter between different states. Expressed in form of a few fundamental principles, it describes the exchange of work and heat of systems with their environment and establishes which processes occur spontaneously. While these laws hold exactly at the macroscopic scale, for small system thermal fluctuations are a dominating factor that cannot be neglected in order to understand significance of dissipation and irreversibility at the nanoscale. Recently important progress has been made in this direction and our understanding of small systems has been changed fundamentally by the discovery of a series of exact relations describing the statistics of fluctuations in non-equilibrium systems. Collaborating with experimentalists from the ETH Zürich, we study these phenomena with levitated nanoparticles in laser traps, which can be modulated to drive the system away from equilibrium. In recent studies, we have analyzed heat fluctuations during relaxation from non-equilibrium states and have investigated the Kramers turnover in a bistable trap.
C. Dellago and G. Hummer, “Computing equilibrium free energies using non-equilibrium molecular dynamics”, Entropy 16, 41-61 (2014).
J. Gieseler, R. Quidant, C. Dellago, and L. Novotny, „Dynamic Relaxation of a Levitated Nanoparticle from a Non-Equilibrium Steady State“, Nature Nanotech. 9, 358-364 (2014).
L. Rondin, J. Gieseler, R. Quidant, C. Dellago, and L. Novotny, "Direct Measurement of Kramers' Turnover with a Levitated Nanoparticle", Nature Nanotech. 12, 1130-1133 (2017).

Nanomaterials
During the last decades tremendous progress has been made in creating novel materials with designed properties and functions based on the unique properties of nanoscale building blocks. The large surface to volume ratios of such systems with sizes in the nanometer range, occupying a middle ground between the macroscopic and the molecular scale, lead to features that can dramatically differ from those of the corresponding bulk materials. For instance, the melting temperatures, band gaps and radiative rates of semiconductor nanocrystals depend sensitively on system size, making these objects interesting for a variety of technological applications ranging from photovoltaics to electronics and biomedicine. In our group, we use molecular dynamics and Monte Carlo simulations combined with enhanced sampling methods to study the properties of a variety of nanomaterials. Our investigations range from pressure induced structural transition in semiconductor nanocrystals and water conduction through carbon nanotube to the irradiate graphene layers and cation exchange.
M. Burian, C. Karner, M. Yarema, W. Heiss, H. Amenitsch, C. Dellago, and R. T. Lechner, "Shape induced orientation phase within 3D nanocrystal solids", Adv. Mat. 30, 1802078 (2018).
L. Frechette, C. Dellago, P. L. Geissler, “Consequences of lattice mismatch for phase equilibrium in heterostructured solids”, Phys. Rev. Lett. 123, 135701 (2019).
C. Karner, C. Dellago, and E. Bianchi, “Design of patchy rhombi: from close-packed tilings to open lattices”, Nano Lett. 19, 7806 (2019).